

Abstract
Recent studies have brought to light additional experimental information, namely, that the MeCP2 protein complex is not only capable of associating with members of the ATPase-dependent bromodomain family, but also found on nonmethylated genomic sequences. These unexpected results are indicative of a multifunctional role for MeCP2, more importantly; our view of the molecular mechanisms that regulate gene activity may not be necessarily distinguishable. Depolarized mouse neuronal cortical cells were examined for increased Slc6a2 mRNA synthesis, changes in CpG methylation status using bisulfite sequencing, and binding of MeCP2 and Smarca2 on the Slc6a2 promoter sequence by chromatin immunopurification strategies. Increased Slc6a2 gene expression in response to membrane depolarization was strongly correlated with the dissociation of MeCP2 and Smarca2 complex on the unmethylated gene. We identified that gene expression in neuronal cortical cells involves increased histone hyperacetylation on the Slc6a2 promoter, which is commensurate with the recruitment of SP1 and RNA Polymerase II and is inversely correlated with H3K9 trimethylation. We hypothesize that the MeCP2 corepressor is capable of associating with multiple forms of SWI/SNF to remodel chromatin for important regulatory roles. The results of our experiments indicate that these proteins are asymmetrically bound to chromatin independent of DNA methylation and not inevitably diametrically opposed. These results now begin to offer a new perspective on the mechanism of Slc6a2 gene regulation.
Introduction
The norepinephrine transporter (NET) regulates the uptake and storage, release and metabolism of noradrenaline in sympathetic nerves. In the CNS, the solute carrier performs critical functions that include neurotransmitter uptake essential in processes such as mood, arousal, learning, blood flow, and metabolism. Although the action of norepinephrine is terminated, by its uptake into noradrenergic neurons by NET, the precise nature of regulation encoded by the Slc6a2 gene remains as controversial as it is equivocal.
The phenotype of reduced neuronal re-uptake of norepinephrine is characteristic of panic disorder and hypertension, and, more recently has been associated with Postural Tachycardia Syndrome (POTS or Autonomic Intolerance) (Jacob et al., 1999; Shannon et al., 2000). Along with the general observation that the causes of orthostatic intolerance compromise diverse pathophysiologies, in one family study the cause of this was traced to a point mutation in the coding region of the Slc6a2 gene resulting in the production of a dysfunctional transporter protein (Shannon et al., 2000). Despite these recent advances, this rare mutation holds little predictive value and there is no direct evidence that sequence polymorphisms identified in the Slc6a2 promoter might account for impaired sympathetic nerve function. Therefore, a fundamental question critical to our understanding of norepinephrine activity is the mechanisms responsible for regulating Slc6a2 gene expression. We have studied the mechanism of Slc6a2 transcriptional inhibition in polarized neurons.
Materials and Methods
Mice and preparation of primary neuronal cultures and depolarization conditions.
The Mecp2tm1Tam mice (Pelka et al., 2006), originally generated on a 129 background, were crossed to C57BL/6 for two generations, followed by F2 intercrosses for 6–8 generations. The mixed background provided hybrid vigor and delayed the onset of symptoms in males enabling more comprehensive behavioral testing. Genotyping of individual pups was determined by PCR of tail DNA using primer sets as previously described (Pelka et al., 2006). Mating of the heterozygous female mutant mice with wild-type males produced hemizygous (−/Y) and heterozygous (+/−) mutant mice and wild-type littermates (+/+, +/Y), which were used as controls. In addition, control C57BL/6 mice were also used in experiments. Ethics approval for animal research was given by local institutional animal ethics committees to conduct these experiments.
Primary cortical neuronal cultures derived from C57BL/6 mice were prepared as described previously (Martinowich et al., 2003; Ciccotosto et al., 2009). Cells were seeded onto poly-d-lysine coated 6-well plates at 4 million cells per well and maintained in a humidified 37°C incubator <5% CO2. For the Mecp2tm1Tam mice, litters from embryonic day 15 mice were placed in separate dishes before the preparation and isolation of cortical neurons to prevent cross-contamination of the genetic background of each individual mouse. Using a modified protocol of cortical isolation for individual mouse embryos (Bellingham et al., 2004), 8–10 million cells were isolated from each embryo and equal numbers of cells were seeded into four separate wells of a six-well plate. Cultures were maintained at 37°C in 5% CO2 for 2 h before the plating medium was replaced with Neurobasal growth medium containing B27 supplements with or without 50 mm KCl.
Quantitative real-time reverse transcription-PCR.
Total RNA was prepared using the Trizol method (Invitrogen) and first strand cDNA synthesis was prepared using M-MLV Reverse Transcriptase (Invitrogen). Quantitative reverse transcription-PCR was performed using Sybr Green Jumpstart Taq Ready Mix (Applied Biosystems).
Chromatin immunoprecipitation primers.
The following chromatin immunoprecipitation (ChIP) primers were used: Mouse Slc6a2 (−2471 to −2263) Forward CAGGTAATGGTCTTGGGCTTG and Reverse CCCTGCTGTTTCATGCCTGT; Mouse Slc6a2 (−2284 to −2263) Forward GAACAGGCATGAAACAGCAGG and Reverse CCATAGGTTCCCGCTCATCA. Mouse Slc6a2 (−1585 to −1382) Forward GCTTTGAGCTTGGAATGTTGC and Reverse GGAGAGCCACGGACAGTTC. Mouse Slc6a2 (−1035 to −832) Forward ACAGTCTGGCCGCAAATAGAG and Reverse CCAGCGTTGTCATACACCCA. Mouse Slc6a2 (−269 to −68) Forward CTCCGAGGCACATCGTGAG and Reverse CTGTGGGTCGGGCTAAAGAG. Mouse Slc6a2 (−150 to +4) Forward CCGCAGGAAACTTGGCATT and Reverse TTTGGAGACTGAGTTGTGACGC. Mouse Slc6a2 (+52 to +149) Forward AGCCCAAAGATCCTGACGTCT and Reverse GCTTGTATGAAAGGAGAAAAGCAG. Mouse H19 Forward CCAGTGCATGTGGTTCGATG and Reverse CCATATCGACCACTGAGGCA. Mouse Rasgrf Forward CACATCCATCCGTGGCTACC and Reverse GACAGCACGGCAGCGAA. Mouse G6pd Forward GGGATAGGGCCTCTCTTGCT and Reverse GCAACTCCCACTCTTCCACCT. Mouse Aprt Forward TCAGCTCCATCCCACAACCT and Reverse CCACCTCCCTAGGCTCGAG.
cDNA primers.
The following cDNA primers were used: Mouse Bdnf Forward CAGGAGTACATATCGGCCACCA and Reverse GTAGGCCAAGTTGCCTTGTCCGT. Mouse Slc6a2 Forward AATCACCCCGGAGAATGAG and Reverse CATTGCCAGGTTCAGATGG. Mouse Gapdh Forward TGAAGCAGGCATCTGAGGG and Reverse CGAAGGTGGAAGAGTGGGAG. Mouse 18S Forward GGCGTCCCCCAACTTCTTA and Reverse GGGCATCACAGACCTGTTATTG.
Bisulfite primers.
The following bisulfite primers were used: Mouse Slc6a2 BIS – Forward TGAGATGGTGTTAGTTAGGTTTT and Reverse CACTCTAAAAAATACAACCACTCAC.
Bisulfite sequencing.
DNA bisulfite conversion was performed following the instructions recommended by the supplier (Millipore Bioscience Research Reagents). One microgram of DNA was used in the conversion reaction, and the DNA was eluted from the carrier by heating it to 60°C in Tris-EDTA buffer and stored at −20°C for PCR amplification. DNA was cloned using the topoisomerase TA vector (Invitrogen), and blue colonies were selected and grown in LB broth. Plasmid was purified using a mini prep kit (Promega), and DNA was sequenced using ABI PRISM Big dye 3.1 Terminator (PE-Applied Biosystems).
Protein immunoprecipitation.
Nuclear extracts prepared from whole mouse brains (Pel-Freez Biologicals) were dounce homogenized, then resuspended in low-salt buffer (10 mm HEPES, 15 mm KCl, 0.2 mm EDTA, 0.1 mm EGTA, 0.5 mm DDT, 0.5 mm spermidine, 0.15 mm spermine, 0.075% NP40) for 30 min at 4°C. Nuclei were isolated by mixing 1 part sucrose buffer (50 mm HEPES, 10 mm KCl, 0.2 mm EDTA, 0.1 mm EGTA, 0.5 mm DDT, 0.5 mm spermidine, 0.15 mm spermine, 75% sucrose) with 2 parts low-salt homogenate and centrifugation at 8000 × g for 30 s. The nuclear extracts were recovered by centrifugation for 15 min at 18,000 × g, and then snap frozen and stored at −80°C. All buffers contained complete protease inhibitor cocktail tablets (Roche Diagnostics). For each immunoprecipitation, 150 μg of nuclear extract containing 1% BSA was precleared twice (2 × 45 min), using 20 μl packed volume protein A Sepharose beads (GE Healthcare). Precleared extracts were incubated for 1 h with antibody to MeCP2 (Sigma), Smarca2, or NFκB-p65 (Abcam), before adding 20 μl packed volume of protein A Sepharose beads blocked with 1% BSA and a further 2 h incubation at 4°C. Bead complexes were washed 5 times with 1 × PBS, 1% Triton X-100 solution, before boiling in 40 μl elution buffer (0.1 m glycine pH 2.5, 50 mm DTT). Precipitated proteins were size-fractionated by SDS-PAGE, transferred to PVDF membrane, and analyzed for Smarca2 (Abcam) by Western blotting using standard techniques.
Chromatin immunopurification and PCR.
Chromatin was sheared for 15 min (30 s on/off cycle) using a Diagenode sonicator to an average length of 700–300 bp in length. Soluble chromatin was immunopurified using MeCP2, Smarca2, H3 unmodified, hyperacetylated H3K9/K14 (antibody recognizes both K9 and/or K14 acetylation), H3K9m3, SP1 and RNA Pol II antibodies, washed, and recovered by elution with 1% SDS, 0.1 m NaHCO3. Purified DNA was subject to PCR using amplimers specific to mouse Slc6a2, Bdnf, Aprt, G6pd, Rasgrf1, and the H19 loci. Cycling parameters on a Gene Amp PCR System (Applied Biosystems) were as follows: 95°C for 4 min followed by 40 cycles of 95°C for 1 min, 58°C for 1 min, and 72°C for 1 min and an extension at 72°C for 10 min. PCR samples were size fractionated using 8% PAGE in 1×TBE buffer, stained with ethidium bromide for 30 min. Gel analyses were performed with the multi-image light cabinet and analyzed using Chemi imager 4400 low light imaging system version 5.1 software (Alpha Innotech).
Quantitative real time PCR.
Quantitation of DNA extracted from ChIP was performed by real time PCR. The data were analyzed by the 7500 Fast Software using Fast Sybr Green ready mix (Applied Biosystems). Quantification was performed using the comparative cycle threshold method and is reported as the n-fold difference in antibody-bound chromatin against the input DNA.
Results
Depolarized cortical neurons show increased Bdnf mRNA levels
We have studied the mechanism by which cytosine methylation might contribute to inhibit transcription of the endogenous Slc6a2 gene in neurons. Recent experimental evidence indicates that membrane depolarization of neuronal cortical cells is associated with specified changes in DNA methylation and this is correlated with the reexpression of the brain-derived neurotrophic factor (Bdnf) gene (Chen et al., 2003; Martinowich et al., 2003). As with methylation-mediated gene silencing, the emerging picture is one of remarkable regulatory complexity, indicating that the MeCP2 corepressor complex is associated with changes in chromatin remodeling activity and Bdnf transcription critical for neural plasticity (Klein et al., 2007).
To determine whether mouse Bdnf mRNA levels had increased following membrane depolarization in cultured neuronal cortical cells, we sought to reproduce recent experimental findings that have indicated that Bdnf gene expression involves methylation-mediated changes involving the release of the MeCP2 corepressor complex from its promoter sequence (Chen et al., 2003; Martinowich et al., 2003). These experiments were made feasible by incubating wild-type mouse cortical neuronal cultures with 50 mm KCl for 72 h. Transcription of Bdnf had significantly increased in neurons after depolarization (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), and these results were compatible with the idea of increased synthesis of Bdnf mRNA levels in neurons following membrane depolarization.
Release of the MeCP2 associated regulatory complex from the activated Bdnf promoter
Soluble chromatin immunopurifications have been used to exemplify that chromatin remodeling activity is associated with the precipitous release of MeCP2 corepressor complex to show that the loss of DNA methylation is critical in the activation of gene transcription (El-Osta et al., 2002). The importance of this mechanism in regulating Bdnf gene expression has recently been reinforced by studies using depolarized neurons (Chen et al., 2003; Martinowich et al., 2003). We therefore tested the central postulate that the MeCP2 associated Smarca2 complex (Harikrishnan et al., 2005, 2006) is linked with the dissociation of the complex from the Bdnf promoter after depolarization. We prepared soluble chromatin fractions and performed ChIPs using antibodies that specifically recognize the MeCP2 and Smarca2 (Brm) proteins, and assessed changes in their association with the Bdnf promoter. Chromatin extractions from depolarized neuronal cortical cells revealed that there was a significant release of MeCP2 and Smarca2 from the Bdnf promoter (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To assess the specificity of the MeCP2-Smarca2 proteins, we also examined the binding of these determinants on the Aprt and G6pd genes, including, the imprinted Rasgrf1 and H19 loci. Binding at these sites was unremarkable, indicating specificity of MeCP2 and Smarca2 on Bdnf chromatin. Together, the experimental data are consistent with the proposed model that the MeCP2 regulatory complex is dissociated from the Bdnf gene following depolarization of postmitotic neurons, which is mediated by reductions in DNA methylation of the promoter, and this is inversely correlated with changes in Bdnf mRNA levels (Chen et al., 2003; Martinowich et al., 2003).
Activation of Slc6a2 gene expression in depolarized neurons
Having determined that the data are consistent with the indirect model of transcriptional repression, which is associated with the recruitment of MeCP2, we postulated that the expression of the Slc6a2 gene, which also has a prominent CpG island, could be regulated by depolarization in neuronal cultures. If this explanation were correct we reasoned that DNA methylation inhibits transcription indirectly by the tight interaction of the MeCP2 corepressor to Slc6a2 chromatin, and, that increased norepinephrine mRNA levels in response to membrane depolarization would be strongly correlated with the dissociation of the MeCP2 complex. Our experimental results indicate that the Slc6a2 gene is activated in depolarized mouse neuronal cortical cells (Fig. 1a).

Activation of Slc6a2 gene expression in depolarized mouse neuronal cortical cells. a, Slc6a2 mRNA levels in response to membrane depolarization in wild-type mouse cortical neuronal cultures. Slc6a2 gene expression was assessed by quantitative realtime reverse transcription PCR and normalized against the expression of the endogenous 18S gene. Samples were analyzed in triplicate and the data are presented as means ± SEM. b, Schematic CpG plot of the mouse Slc6a2 gene showing the CpG island relative to the transcription initiation start site at +1. c, Bisulfite sequencing analysis of 46 CpG sites of the Slc6a2 gene sequence in wild-type and depolarized neuronal cultures. More than 12 clones from the wild-type cells were assessed for sequencing from 2 separate cultures. To determine the significance of CpG methylation in depolarized neuronal cultures, 11 clones were assessed from two individual cultures.
Slc6a2 promoter is hypomethylated
To specifically determine changes in the CpG island of the Slc6a2 gene (Fig. 1b), we used bisulfite sequencing of individual clones derived from wild-type and depolarized neuronal cultures. As can be seen in Figure 1c, bisulfite-sequencing analysis performed on 46 CpG sites (−499 to +81) of the gene indicates the Slc6a2 promoter sequence is unmethylated in control and depolarized neurons. If depolarization mediates a release of the MeCP2 corepressor from gene sequences because of specific demethylating events such as those observed with the Bdnf gene, then our results would argue against the involvement of the corepressor on unmethylated Slc6a2 sequences. The correspondence of neuronal depolarization alleviating the suppression of Slc6a2 transcription was also strongly correlated with Bdnf gene activity; yet, demethylation could not explain the reactivation of Slc6a2 gene expression, unless transcriptional output was related to the ability of the MeCP2 complex to associate with unmethylated sequences (Yasui et al., 2007). In fact, the importance of MeCP2 binding to unmethylated gene sequences was recently reinforced in the mouse hypothalamus (Chahrour et al., 2008), which was first suggested for homologous MeCP2 proteins (Weitzel et al., 1997). However, other evidence indicates that MeCP2 may in fact contribute to gene expression not involving the covalent addition of methyl groups at the 5′ position of cytosine within CpG dinucleotides (Yasui et al., 2007), and this was later confirmed in vivo (Chahrour et al., 2008). Further evidence suggesting that unmethylated sequences could be involved in recruiting MeCP2 and mediating gene suppression are derived from recent observations that MeCP2 specifically binds unmethylated DNA and alters chromatin structures (Galvão and Thomas, 2005). The existing literature suggests a multifunctional role for MeCP2 in regulating gene activity. These recent studies have also brought to light additional experimental information and the intriguing possibility that the MeCP2 regulatory complex could be involved in the repression of unmethylated genes (Cukier et al., 2008). In view of these observations, the emerging picture is one of remarkable complexity for MeCP2. In this context, and, consistent with striking recent experimental observations, exclusivity of MeCP2 with methylation-mediated gene silencing could be considered overly reductionist.
Activation of Slc6a2 gene expression is correlated with the dissociation of MeCP2 and Smarca2 regulatory complex on the unmethylated Slc6a2 gene sequence
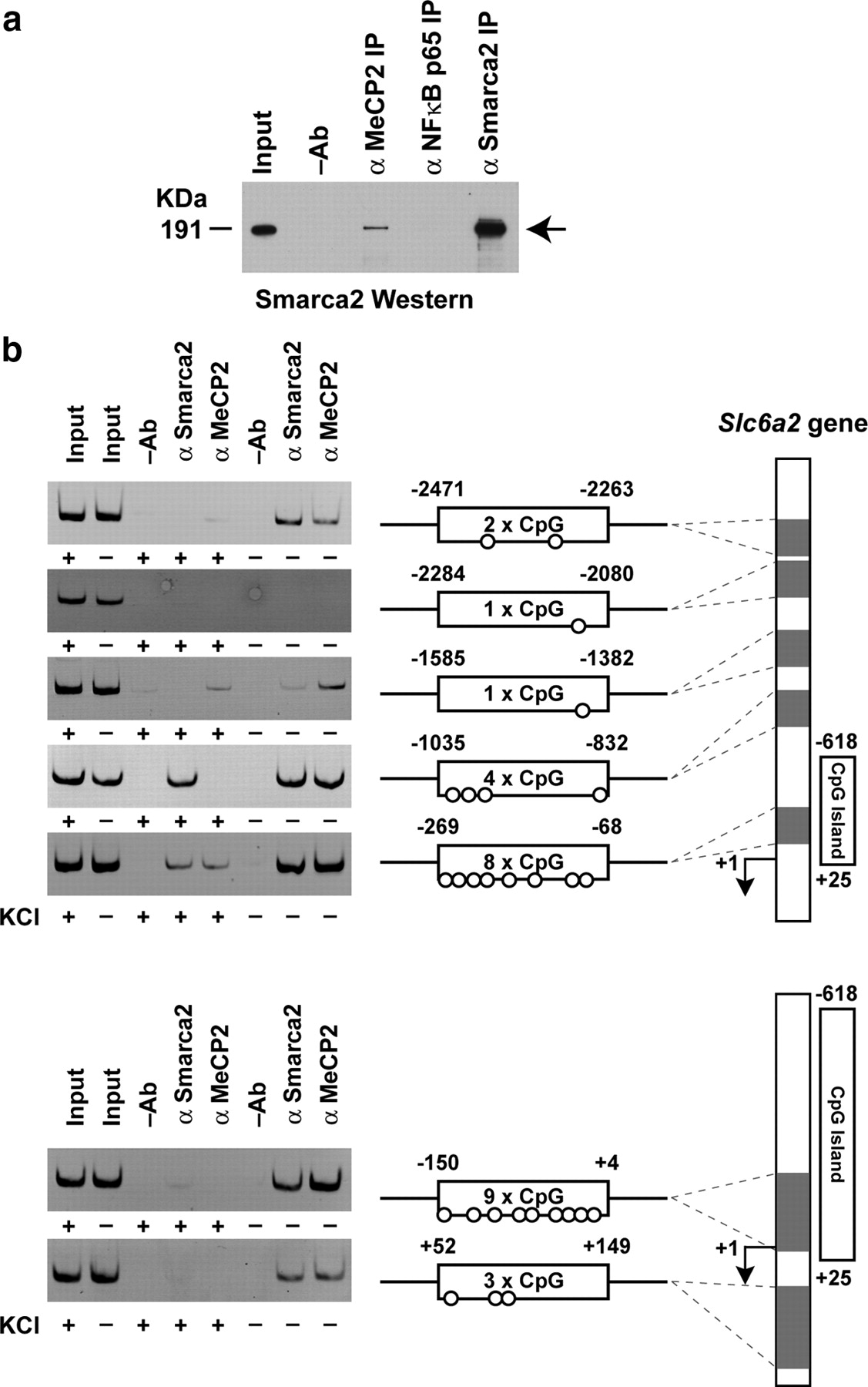
To be able to study the hypothesis that the MeCP2-Smarca2 complex could be associated with the unmethylated Slc6a2 sequence, we first determined whether these proteins participate by coimmunoprecipitation assays. Consistent with recent evidence that MeCP2 associates with the large molecular weight ATPase-remodeling complex (Harikrishnan et al., 2005, 2006), experiments performed on nuclear extracts derived from mouse brain demonstrate that MeCP2 coimmunoprecipitates the Smarca2 subunit of SWitch/Sucrose Non-Fermentable (SWI/SNF) (Fig. 2a). To establish whether the MeCP2 and Smarca2 proteins associate with the Slc6a2 gene, we performed chromatin immunopurification assays. To do this we deliberately designed a chromosomal walk that would allow us to assess and compare protein distribution with differing cytosine-guanine dinucleotide densities of the Slc6a2 gene. The ChIP walk examined the CpG island sequence from −2471 to +4, including the +52 to +149 region of the Slc6a2 promoter in wild-type and depolarized neurons. Chromatin fractions were specifically immunopurified for these endogenous proteins using the MeCP2 and Smarca2 antibodies; we then assessed the level of determinant enrichment with the unmethylated Slc6a2 gene. ChIP analyses from mouse nondepolarized neuronal cultures revealed that MeCP2 and the Smarca2 regulatory proteins are tightly associated with sequences −150 to +4, −269 to −68, and −1035 to −832 of the Slc6a2 gene, which correspond to 9, 8, and 4 CpG sites, respectively (Fig. 2b). It is noted that differences in protein binding are correlated with CpG density, and, therefore it is conceivable that differing affinities of the Slc6a2 gene sequences associate with the MeCP2 and Smarca2 proteins. Interestingly, after neuron depolarization MeCP2 association was clearly distinguishable on all tested sequences, and in regions −150 to +4 and −1035 to −832 we could barely detect the MeCP2 regulatory protein on the Slc6a2 gene. The release of the MeCP2 and Smarca2 proteins was demonstrable on all tested sites of the Slc6a2 gene, except for region −1035 to −832, showing incommensurable binding for Smarca2. To test the binding specificity of the MeCP2 and Smarca2 regulatory complex we also assessed the association of these determinants on the unrelated Aprt gene, which confirmed the specificity of MeCP2 and Smarca2 binding (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Together, these results show a close correspondence between Slc6a2 gene activation in depolarized neurons and the dissociation of the MeCP2 and Smarca2 regulatory complex.

Activation of Slc6a2 gene expression in depolarized mouse neuronal cortical cells is strongly correlated with the dissociation of MeCP2 and Smarca2 regulatory complex on the unmethylated Slc6a2 gene sequence. a, Association of MeCP2 with Smarca2 was determined by coimmunoprecipitation assay of mouse brain nuclear extracts. Coimmunopurified Smarca2 was assessed by protein immunoblotting. Antibody that recognizes NFκB nuclear protein was used in the IP to assess assay specificity. b, Chromatin immunopurifications of MeCP2 and Smarca2 binding on the Slc6a2 gene after membrane depolarization in mouse cortical neuronal cultures. Schematic representation of the upstream and proximal promoter sequence mapped by chromosomal walking is shown on the right panel. Shaded regions representing the amplicons relative to the transcription initiation start site (+1) are shown, including the CpG density of each amplicon. Soluble chromatin input and no antibody immunopurifications (−Ab) were also assessed as controls for assay specificity.
The release of MeCP2 is inversely correlated with increased histone hyperacetylation on the unmethylated Slc6a2 gene sequence in depolarized neurons
MeCP2 is associated with the suppression of gene activity, which is thought to be associated with distinct regulatory complexes (Harikrishnan et al., 2006; Hu et al., 2006) in specific cell types and can be associated with histone deacetylase activity (Jones et al., 1998; Nan et al., 1998). We therefore examined whether the suppression of Slc6a2 gene activity by MeCP2 regulatory complex was inversely linked with histone modification on the promoter, specifically, whether depolarized neurons show increased acetylation of histones on the Slc6a2 gene. If this explanation was correct, we reasoned that the specific release of the regulatory MeCP2 complex from the Slc6a2 promoter would increase the acetylation of histone H3K4/K14 in neuronal cortical cells after membrane depolarization. To test this hypothesis, we performed chromatin immunopurifications of MeCP2 and histone H3K9/K14 hyperacetylation on the Slc6a2 gene in wild-type and depolarized mouse cortical cells. ChIP analyses from mouse depolarized neuronal cultures show that MeCP2 and H3K9/K14 acetylation are inversely correlated on the Slc6a2 promoter, and this is clearly seen with sequences −1585 to −1382, −1035 to −832, and −269 to −68 of the promoter (Fig. 3a). To determine the degree of MeCP2 binding and changes in histone H3K9/K14 acetylation and trimethylated H3K9, we assessed the association of these determinants on the Slc6a2 promoter by quantitative real time PCR and clearly observe increased hyperacetylation on −1035 to −832 (Fig. 3b) and −269 to −68 of the promoter with reciprocal reductions in MeCP2 binding on the Slc6a2 gene in depolarized neurons (Fig. 3c). ChIP experiments show parallel associations between MeCP2 that indicate H3K9m3 is also significantly reduced on the Slc6a2 gene in depolarized neurons. These experiments gave identical results to independent experiments examining MeCP2 and Smarca2 promoter walk. Soluble chromatin input and an equivalent amount of IgG isotype antibody were also assessed as controls for assay specificity. The results to these experiments suggest a reciprocal exchange of the MeCP2 coregulatory complex following membrane depolarization to a less suppressive chromatin structure that is associated with enhanced hyperacetylation of the Slc6a2 gene sequence.

Activation of Slc6a2 gene expression in depolarized mouse neuronal cortical cells is correlated with increased hyperacetylation of histone H3K9/K14 and the release of the MeCP2 regulatory complex with reduced H3K9m3 on the unmethylated sequence. a, Chromatin immunopurifications of MeCP2 and histone H3K9/K14 hyperacetylation on the Slc6a2 gene after membrane depolarization in mouse cortical neuronal cultures. Schematic representation of the upstream and proximal promoter sequence mapped by chromosomal walking is shown on the right panel. Shaded regions representing the amplicons relative to the transcription initiation start site (+1) are shown, including the CpG density of each amplicon. Soluble chromatin input and an equivalent amount of IgG isotype antibody were also assessed as controls for assay specificity. b, Quantification of MeCP2, acetylated H3K9/K14, and trimethylated H3K9 chromatin immunopurifications on the Slc6a2 promoter region −1035 to −832. Samples were analyzed in triplicate and the data are presented as means ± SEM. c, Quantification of MeCP2, acetylated H3K9/K14, and trimethylated H3K9 chromatin immunopurifications on the Slc6a2 promoter region −269 to −68. Samples were analyzed in triplicate and the data are presented as means ± SEM.
Activation of Slc6a2 gene expression in depolarized mouse neuronal cortical cells involves the recruitment of SP1 and RNA Polymerase II and is inversely correlated with H3K9 methylation
Having established in depolarized neurons that increased gene expression is associated with the release of the MeCP2 complex that is consistent with enhanced H3K9/K14 hyperacetylation on the Slc6a2 promoter, we assessed whether this change in chromatin structure could result in the recruitment of transcriptional coactivators to the Slc6a2 gene.
We hypothesized that potential SP1 binding sites of the mouse Slc6a2 promoter could be directly involved in this transcriptional event (Kim et al., 1999). To test whether the alleviation of transcriptional suppression was associated with increased SP1 binding, independent experiments using neuronal cortical cultures were performed and we assessed the Slc6a2 (−150 to +4) promoter for reciprocal exchanges of these determinants by chromatin immunopurification (Fig. 4a). Because transcriptionally active genes have been shown to exhibit increased chromatin accessibility mediated in part by histone modification (enhanced H3K9/K14 acetylation and reduced tri-methylation of lysine 9; H3K9m3), we also examined these specific modifications as well as the recruitment of RNA Pol II as a measure of increased gene activity. Depolarized cortical cells show clear enrichment of SP1 binding to sequences −150 to +4 (relative to the transcription start site), which are putative SP1 elements of the Slc6a2 promoter (Fig. 4b). Since increased SP1 binding and Slc6a2 gene expression are commensurate and inversely correlated with MeCP2 release, we directly assessed RNA Pol II. ChIP analyses indicate a consistent increase in the association of the RNA Pol II on the promoter, which is suggesting that Slc6a2 transcription, was indeed increased in response to membrane depolarization (Fig. 4b). Our results also indicate that increased H3K9/K14 hyperacetylation was inversely associated with H3K9m3 and MeCP2 binding on the Slc6a2 gene sequence. In each of these experiments, soluble chromatin input and an equivalent amount of IgG isotype antibody were assessed confirming assay specificity.

Activation of Slc6a2 gene expression in depolarized mouse neuronal cortical cells involves the recruitment of SP1 and RNA Polymerase II and increased hyperacetylation of histone H3K9/K14 for the exchange and the dissociation of the MeCP2 regulatory complex on the unmethylated Slc6a2 promoter. a, Schematic representation of the upstream and proximal promoter sequence mapped by chromatin immunopurification. The shaded region represents the amplicon relative to the transcription initiation start site (+1), including the CpG density of the amplicon (left). Chromatin immunopurifications of MeCP2, histone H3K9/K14 hyperacetylation, SP1 and RNA Polymerase II on the Slc6a2 gene after membrane depolarization in mouse cortical neuronal cultures (right). b, Quantification of MeCP2, acetylated H3K9/K14, H3K9m3, SP1, and RNA Polymerase II chromatin immunopurifications on the Slc6a2 promoter region −150 to +4. Samples were analyzed in triplicate and the data are presented as means ± SEM. Soluble chromatin input and an equivalent amount of IgG isotype antibody were also assessed as controls for assay specificity.
Neuronal cells cultured from Mecp2 mutant mice displayed increased Slc6a2 expression
The experimental results we have observed indicate that the transcriptional competence of the Slc6a2 gene is directly associated with changes in histone acetylation and the recruitment of transcription factor SP1 and RNA Pol II in depolarized cortical cell cultures, if MeCP2 is also associated with the suppression of gene activity, then we would expect that neuronal cells isolated from Mecp2 mutant brains to show increased expression of this gene. To directly test this idea we isolated neuronal cortical cells from brains (embryonic day 15) of wild-type Mecp2 (+/Y) and Mecp2 mutants (−/Y) (Pelka et al., 2006). Depolarized wild-type (+/Y) cells show increased Slc6a2 gene expression that is comparable to that observed for Mecp2 mutant (−/Y) cells that were not stimulated with potassium chloride (n = 4 pairs of animals) (Fig. 5). The results to these experiments indicate that Slc6a2 gene expression levels in MeCP2-deficient neuronal cells were not significantly different in depolarized and unstimulated cells, suggesting a role for MeCP2 in the regulation of Slc6a2 gene expression. These results are also consistent with the role for MeCP2 in the expression of gene activity in response to membrane depolarization (Chen et al., 2003; Chang et al., 2006). Together, the close correlation between MeCP2 and Smarca2 affinity with gene repression and the release of this regulatory complex with the exchange and recruitment of coactivator determinants following membrane depolarization indicate a functional role for the MeCP2 complex with the transcriptional activity of the Slc6a2 gene (Fig. 6).

MeCP2 regulates Slc6a2 gene expression. Mouse neuronal cells isolated from brains (embryonic day 15) of wild-type Mecp2 (+/Y) and Mecp2 mutants (−/Y) were cultured for +3 days in vitro and gene expression was assessed after depolarization with potassium chloride. Slc6a2 gene expression was assessed by quantitative real time reverse transcription PCR and normalized against the expression of the endogenous GAPDH gene. Samples were analyzed in triplicate and the data are presented as means ± SEM.
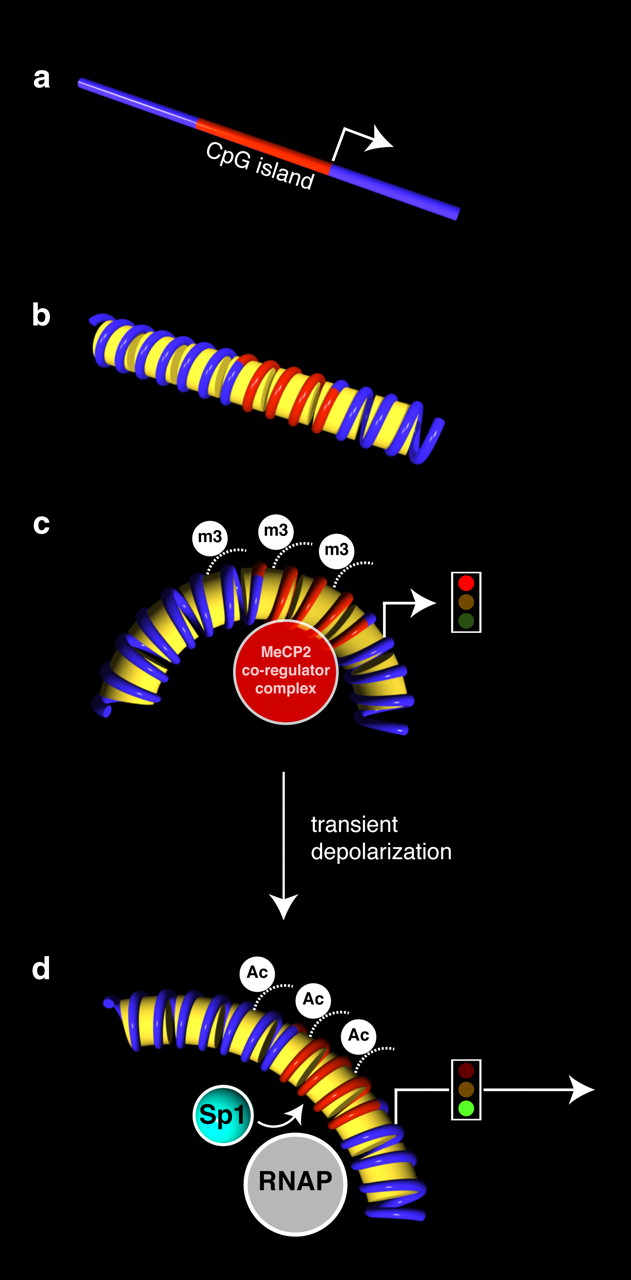
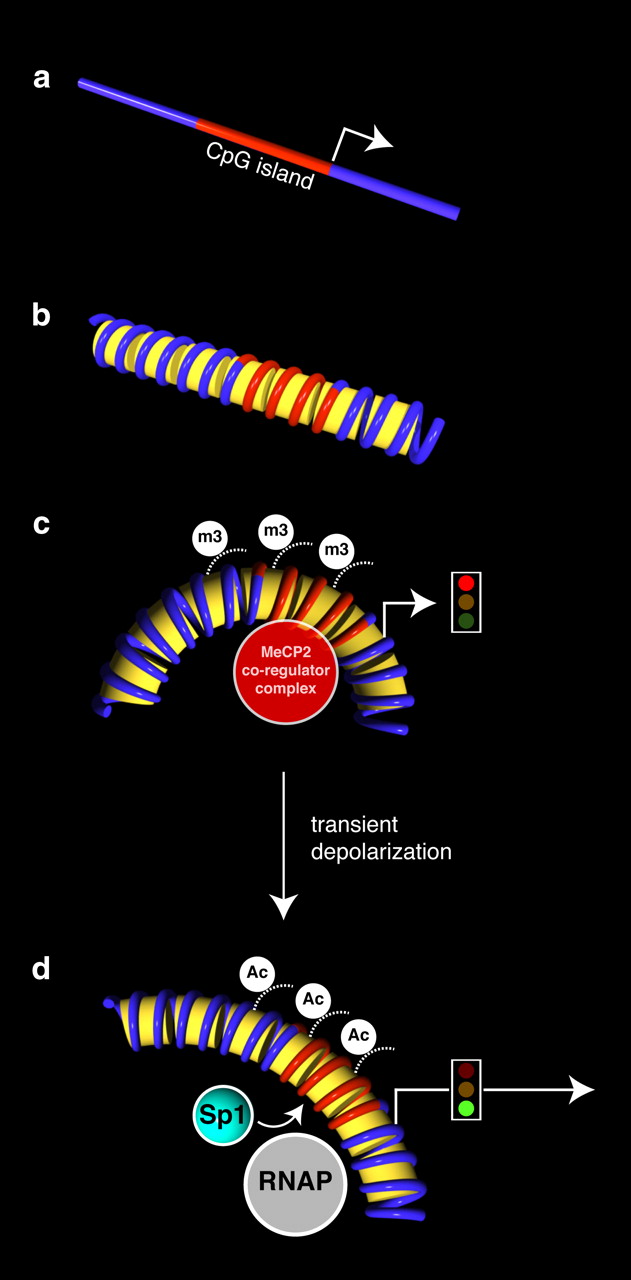
Hypothetical model of Slc6a2 gene reexpression following membrane depolarization. a, Schema of the mouse Slc6a2 gene illustrating the unmethylated CpG island and the transcription initiation start site (arrowhead). b, Conceptualized Slc6a2 chromatin structures are shown. c, The activation of Slc6a2 gene expression is correlated with the dissociation of MeCP2 and Smarca2 regulatory complex on the unmethylated Slc6a2 gene sequence. Gene suppression is correlated with the association of the MeCP2 regulatory complex including Smarca2 and H3K9m3 (m3) histone modification. d, Following membrane depolarization and increased Slc6a2 expression, specific molecular events using chromatin immunopurification strategies identified the recruitment of SP1 and RNA Polymerase II, which are inversely correlated with H3K9 methylation. The active gene is hyperacetylated (Ac) at the Slc6a2 promoter.
Discussion
In this study, we have demonstrated that the activation of the hypomethylated Slc6a2 gene is correlated with the release of the MeCP2 associated Smarca2 regulatory complex. We identified that the suppression of Slc6a2 transcription can be alleviated with membrane depolarization in neuronal cortical cells and the coincident recruitment of SP1 and RNA Pol II on the promoter of Slc6a2. The dissociation of MeCP2 complex was also inversely associated with increased H3 hyperacetylation. We also show that neuronal cells cultured from MeCP2 mutant mice display increased Slc6a2 gene expression. The close correspondence between the MeCP2 associated Smarca2 regulatory complex and the association on the unmethylated and suppressed Slc6a2 sequence is not easily explained by chemical modification of the promoter as determined by bisulfite sequencing. This latter result was not anticipated unless MeCP2 directly binds to unmethylated CpG sequences of the Slc6a2 promoter. Together, these experimental results suggest that the MeCP2 coregulatory complex confers the suppression of Slc6a2 gene activity.
These experimental results contrast with those for Bdnf gene transcription, indicating that membrane depolarization is directly linked with increased transcription conferred by the dissociation of the MeCP2 associated Smarca2 complex from the Bdnf promoter. These transcriptional properties identified on the Bdnf promoter in neuronal cells are consistent with the concept that Bdnf gene activity is inversely correspondent with changes in genomic methylation and the release of the corepressor complex from Bdnf sequence sites (Chen et al., 2003; Martinowich et al., 2003). Although we cannot exclude that association of the MeCP2 corepressor complex could be mediated by other regulatory proteins, we hypothesize that Slc6a2 gene activity is independent of sequence methylation. These results at present cannot be explained by the precedent model that has for so long relied on the paradigm of methylation-mediated silencing. These experimental results are unanticipated but not unprecedented. MeCP2 has also been distinguished with the expression of gene activity, furthermore raising the intriguing possibility of a multifunctional role for MeCP2 (Cukier et al., 2008). These studies present a complicated and even contradictory set of experimental data on the binding preferences for MeCP2 and its regulatory roles in transcription (Weitzel et al., 1997; Yasui et al., 2007; Chahrour et al., 2008).
The MeCP2 molecule has only a 2–3-fold preference for methylated over unmethylated sequences (Hite et al., 2009). Although our results highlight an association between the MeCP2 protein complex and the Slc6a2 gene, it is not clear whether this binding is mediated by the N-terminal region of the MBD or another region of the MeCP2 protein. MeCP2 has the ability to form chromatin complexes mediated by the methylated DNA binding domain (Georgel et al., 2003; Adams et al., 2007). The results of these experiments suggest that the MBD and TRD domains of the MeCP2 protein may not only function alone, but, could also allow combination of the aforementioned domains to act together (Adams et al., 2007). Although the precise nature of DNA recognition remains controversial, recent studies have identified methylation-independent DNA binding region in the N-terminal region of MeCP2, and, an alternative chromatin interaction region in the C terminus suggesting methylation-dependent and -independent interactions (Nikitina et al., 2007). Clearly, future work to specifically distinguish these regions of the MeCP2 protein will further clarify how these interactions are mediated on the chromatin template. Furthermore, we cannot exclude that in the course of recognizing and binding unmethylated sequences, MeCP2 may be indirectly recruited to the Slc6a2 promoter by intermediary protein-protein associations.
The norepinephrine transporter plays a central role regulating noradrenaline in the nervous system, and, the identification of the specific regulatory complexes under the control of diverse epigenetic pathways that underlie Slc6a2 gene deregulation could be critical in providing new therapeutic targets associated with various pathophysiologies that include neurodegenerative, psychiatric, and cardiovascular disorders. Recent studies have also shown that polarized sympathetic neurons show increased norepinephrine expression mediated by catecholamine production (Habecker et al., 2006).
We hypothesize that the MeCP2 protein is capable of associating with multiple forms of SWI/SNF to remodel chromatin for important regulatory functions. The results of our experiments indicate that these proteins are asymmetrically bound to Slc6a2 chromatin independent of DNA methylation, and, binding of the MeCP2 complex to methylated sequences are not inevitably diametrically opposed as previously postulated. These results now begin to offer a new perspective on the mechanism of norepinephrine transporter gene regulation and the intriguing possibility of demonstrable relevance in various pathophysiologies to understanding the faulty neuronal norepinephrine re-uptake phenotype.
Footnotes
-
We acknowledge grant and fellowship support from the National Health and Medical Research Council of Australia.
- Correspondence should be addressed to Assam El-Osta, Epigenetics in Human Health and Disease Laboratory, Baker IDI Heart and Diabetes Institute, 75 Commercial Road, The Alfred Medical Research and Education Precinct, Melbourne, VIC 3004, Australia. assam.el-osta{at}bakeridi.edu.au





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}