

Abstract
Synaptic plasticity rules widely determine how cortical networks develop and store information. Using confocal imaging and dual site focal synaptic stimulation, we show that basal dendrites, which receive the majority of synapses innervating neocortical pyramidal neurons, contain two compartments with respect to plasticity rules. Synapses innervating the proximal basal tree are easily modified when paired with the global activity of the neuron. In contrast, synapses innervating the distal basal tree fail to change in response to global suprathreshold activity or local dendritic spikes. These synapses can undergo long-term potentiation under unusual conditions when local NMDA spikes, which evoke large calcium transients, are paired with a “gating molecule,” BDNF. Moreover, these synapses use a new temporal plasticity rule, which is an order of magnitude longer than spike timing dependent plasticity and prefers reversed presynaptic/postsynaptic activation order. The newly described plasticity compartmentalization of basal dendrites expands the networks plasticity rules and may support different learning and developmental functions.
Introduction
Synaptic plasticity rules widely determine how cortical networks store information. Presently, the best-studied set of plasticity rules is spike timing-dependent plasticity (STDP), in which plasticity is mediated by the global postsynaptic signal of back-propagating action potentials (BAPs) (Paulsen and Sejnowski, 2000; Sjostrom et al., 2002; Dan and Poo, 2004). Previous studies from pyramidal neurons revealed that active and passive properties vary along apical dendrites giving rise to functional differences between proximal and distal dendritic regions (Schiller et al., 1997; Stuart et al., 1997; Larkum et al., 1999a; Magee, 1999, 2000; Migliore et al., 1999; Johnston et al., 2000; Korngreen and Sakmann, 2000). Plasticity rules may also differ along the dendritic tree. Previous studies reported that STDP rules depend on the site of the synapse along the apical dendritic tree of neocortical pyramidal neurons (Froemke et al., 2005; Letzkus et al., 2006; Sjostrom and Hausser, 2006). In addition, synapses located in distal apical dendrites can use local dendritic spikes as the postsynaptic signal to induce plasticity (Golding et al., 2002).
The dendritic tree of pyramidal neurons is subdivided into the apical and basal arborizations. The majority of synaptic inputs to neocortical pyramidal neurons are received and processed by the thin basal dendrites (Larkman, 1991). Despite the importance of basal dendrites the plasticity rules in these dendrites in general and their location dependence in particular are mostly unknown. We were especially interested in this question because, in contrast to apical dendrites, distal basal dendrites typically support initiation of NMDA spikes that in turn are associated with large local calcium influx (Schiller et al., 2000). As induction of most forms of long-term synaptic plasticity requires calcium influx via activated NMDA-receptor channels (Artola and Singer, 1993; Bliss and Collingridge, 1993; Franks and Sejnowski, 2002; Sjostrom and Nelson, 2002), we hypothesized that NMDA spikes can serve as a local postsynaptic signal for induction of long-term potentiation (LTP), especially in distal basal dendrites, in which these spikes are more easily initiated. Moreover, the unique properties of the NMDA spike, especially with respect to its long duration and highly localized nature (Schiller et al., 2000), are expected to result in different plasticity rules than those in apical dendrites. In this study, we used combined intracellular recordings, calcium imaging, and focal synaptic stimulation in neocortical layer 5 and layer 2–3 pyramidal neurons (Schiller et al., 2000; Polsky et al., 2004) to investigate the plasticity rules along basal dendrites. We report that these rules differ significantly from those reported previously in apical dendrites. Although similar to apical dendrites, synapses innervating proximal basal dendrites undergo LTP in response to pairing EPSPs with BAPs;inputs innervating distal basal dendrites fail to undergo LTP when paired with either BAPs or local spikes and require both initiation of NMDA spikes and a cofactor molecule in the form of BDNF. Moreover, the temporal plasticity rules in distal basal dendrites markedly differ from those reported previously for apical dendrites and those we found in proximal basal dendrites.
Materials and Methods
Slice preparation and electrophysiological recording.
Neocortical parasagittal brain slices from the somatosensory area, 300–350 μm thick, were prepared from 17- to 24-d-old female and male Wistar rats. Whole-cell patch-clamp recordings were performed from visually identified layer 5 and layer 2–3 pyramidal neurons using infrared-differential interference contrast optics. The extracellular solution contained the following (in mm): 125 NaCl, 25 NaHCO3, 25 glucose, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, pH 7.4 at 35 to 36°C. The intracellular solution contained the following (in mm): 115 K+-gluconate, 20 KCl, 2 Mg-ATP, 2 Na2-ATP, 10 Na2-phosphocreatine, 0.3 GTP, 10 HEPES, 0.2 Oregon Green 488 Bapta-1 (OGB-1) or Bapta-6 (OGB-6), pH 7.2. Bicuculline methiodide (1 μm) was added to the extracellular solution. The electrophysiological recordings were performed using Multi-Clamp 700A (Molecular Devices, Foster City, CA), and the data were acquired and analyzed using pClamp 8.2 (Molecular Devices), a homemade software, and Igor (Wavemetrics, Lake Oswego, OR) software. Statistical tests were performed using Excel software (Microsoft, Redmond, WA). Measurements are reported as mean ± SEM. Statistical significance was tested using paired Student's t test.
Focal synaptic stimulation and calcium fluorescence imaging.
Focal synaptic stimulation was performed via a theta-patch pipette located in close proximity to the selected basal dendritic segment guided by the fluorescent image of the dendrite. In these experiments, the neurons were filled with the calcium-sensitive dye (OGB-1 or OGB-6, 200 μm) and the basal dendritic tree was imaged with a confocal system (Fluoview 500; Olympus, Tokyo, Japan) mounted on an upright BX51WI Olympus microscope equipped with a 60× (0.9 numerical aperture; Olympus) water objective. The theta-stimulating electrodes were filled with Alexa Fluor 647. The location of the activated inputs was performed as described previously (Polsky et al., 2004). Briefly, small EPSPs (0.5–1 mV at the soma) were evoked and concomitant calcium imaging was performed to located the active dendritic segment and active dendritic spines. Typically, we observed calcium transients that were restricted to a dendritic segment of 10–20 μm. When a dendrite was activated by two adjacent stimulating electrodes (30–40 μm apart), we monitored the overlap between the two stimulated segments using calcium imaging measurements, as described previously (Polsky et al., 2004).
The extent of the calcium spread was monitored separately for each stimulating electrode. For the electrode that was used to evoke the NMDA spike, the extent of the calcium spread was determined with stimulation intensities just subthreshold to NMDA-spike initiation (once the spike initiated, there was spatial spread of the calcium transient probably caused by secondary activation of voltage-gated calcium channels activated by the large voltage signal). In all experiments, we made sure that the dendritic segments activated by the two stimulating electrodes (as determined by the calcium imaging) did not overlap significantly.
Full images were obtained with a temporal resolution of 1 Hz, and in the line scan mode with a temporal resolution of 512 Hz. Images were analyzed using Tiempo (Olympus), homemade software, and Igor software. Fluorescence changes were quantified as increase in fluorescence from baseline normalized by the baseline fluorescence (ΔF/F as a percentage). The background florescence was subtracted from all measurements before calculation of the ΔF/F. Calcium transients are reported as mean ± SD. For comparison of calcium transients in proximal and distal locations, we used a high concentration (0.5–1 mm) of the calcium sensitive dye OGB-6 and data acquisition was done with a line scan from active dendritic spines.
Destruction of basal branches with laser exposure.
Basal branches were destroyed by 2–3 min laser exposure in the line scan mode with both the 488 and 633 nm lasers. To avoid damage to the soma, the segment damaged was typically 15–25 μm away from the soma. The damage was monitored using fluorescence imaging, with damaged branches appearing inflated, “blebby,” and saturated with calcium.
Glutamate uncaging.
For the uncaging experiments, caged glutamate [4-methoxy-7-nitroindolinyl (MNI)-glutamate; Tocris, San Diego, CA] was photolyzed with a 361 nm UV-laser beam (Enterprise 2; Coherent, Palo Alto, CA) using point scan mode. The caged glutamate (5–10 mm) was delivered locally to a branch using pressure ejection (5–10 mbar) from an MNI-glutamate-containing electrode (2 μm diameter).
Long-term plasticity induction protocols.
EPSPs were evoked at 0.1 Hz before and after the induction protocols. Control EPSPs were acquired for 10–20 min before induction. Stability of the experiments was ensured by monitoring the membrane resting potential and the amplitude of the preinduction control EPSPs. In all experiments included, the resting membrane potential was stable within 2 mV and the preinduction control EPSPs (average of six consecutive EPSPs) showed less than a 20% change. LTP was induced using three different protocols. For pairing NMDA spikes and EPSPs, we typically repeated the pairing 15–30 times at 0.1 Hz. In some experiments, we used only five repetitions or up to 60 repetitions of the induction protocol. In addition we used a theta burst protocol, in which NMDA spikes and EPSPs (2 stimuli at 100 Hz) were paired five times at 5 Hz, and this sequence was repeated five times at 0.1 Hz. In these experiments, the stimulating electrode was positioned mainly at the distal half of the dendrites as spike initiation was easier at these dendritic regions. In some of the experiments (mentioned in the text), BDNF (50 ng/ml in artificial CSF containing 0.1% BSA; Alomone Labs, Jerusalem, Israel) was applied via pressure ejection (10–20 mbar) through a glass pipette (tip diameter of 2 μm) locally to a dendrite of interest. The BDNF was applied throughout the induction period only. For pairing protocols, we used three different induction protocols, the first consisted of 30–60 repetitions at 0.1 Hz of pairing a train of BAPs at 50–150 Hz (evoked by short 5 ms depolarizing somatic current injections) with a single EPSP. The EPSP was evoked 10 ms before the BAP burst. The second protocol consisted of 15 repetitions at 0.05 Hz of pairing a burst of BAPs evoked by a 100 ms depolarizing current injection at the soma with a burst of four EPSPs at evoked at 50 Hz. The EPSP train preceded the BAP burst by 10 ms (Markram et al., 1997b; Sjostrom et al., 2001). The third induction protocol (theta burst stimulation) consisted of pairing a burst of three BAPs evoked by a depolarizing current injection paired with five EPSPs evoked at 100 Hz. This short burst was repeated five times at 5 Hz (theta rhythm), and this sequence was repeated five times at 0.1 Hz (Pike et al., 1999; Golding et al., 2002; Dan and Poo, 2004).
Results
Spike timing-dependent plasticity in proximal and distal basal dendrites
To investigate the rules governing long-term plasticity changes at different locations along basal dendrites, we used the well characterized induction protocol, which consists of pairing EPSPs with BAPs. To differentially activate inputs innervating the proximal and distal portions of basal dendrites, we used focal synaptic stimulation with bipolar theta electrodes combined with confocal fluorescence imaging (Fig. 1A) (Koester and Sakmann, 1998; Schiller et al., 2000; Polsky et al., 2004). Experiments were performed only when focal stimulation evoked well defined calcium transients restricted to a segment of the proximal or distal areas including the identification of active spines. To estimate what fraction of the EPSP originated from the activated dendrite in our experiments, we measured the EPSP amplitude before and after deliberately destroying the activated branch by prolonged intense laser exposure. In these experiments after laser induced destruction of the activated branch, the EPSP amplitude decreased by 80 ± 14% for proximal stimulation and 85.1 ± 11% for distal stimulation (n = 6; p > 0.3). Hence, we concluded that the majority of the voltage originated from the stimulated target basal branch.

Differential response to pairing induction protocol of proximal versus distal synapses in basal dendrites. A, Experimental setup. A layer 5 pyramidal neuron was loaded with OGB-1 (200 μm), and two theta-stimulating electrodes were positioned, one proximal (red, 70 μm from the soma) and one distal (blue, 200 μm from the soma). Calcium transients evoked by small EPSPs (1–2 mV) from proximal (red box) and distal (blue box) regions were collected in the line scan mode. Graphical representation (ΔF/F as a percentage) is shown for three different regions of the line scan marked by the numbers 1–3 (red numbers, proximal; blue numbers, distal). The regions marked by 2 show significant calcium transients, whereas in regions 1 and 3, the calcium transients are markedly diminished. Scale bar, 50 μm. B, LTP was induced using a pairing protocol consisting of a burst of BAPs (top; 4 action potentials; 50 Hz), delivered 10 ms after a single EPSP. Example EPSP traces (average of 15 traces) before (30 min) and after (50 min) induction of the proximal (red) and distal EPSPs (blue) are shown. Note that only the proximally generated EPSPs underwent LTP, whereas the distally generated EPSPs were slightly depressed. C, Peak EPSPs originating from the proximal (red) and distal (blue) were measured simultaneously at the soma before and after induction (black bar, induction period). D, Summary of normalized average data from 22 proximal and 14 distal dendritic locations (red, proximal regions; blue, distal regions) shows strong potentiation at proximal synapses and slight depression at distal synapses. Two main reasons caused the slower time course in the averaged potentiation curve compared with the example presented in C. First, in some neurons, potentiation occurred gradually. Second, there were differences in the timing of the step-like potentiation between different neurons. Error bars represent SEM.
Pairing a train of BAPs (four action potentials, 50–150 Hz) with an EPSP generated at the proximal half of basal branches evoked LTP (Fig. 1). Under these conditions, most neurons examined (17 of 22; 77%) were potentiated and the EPSP amplitude increased to an average of 185 ± 32% and 215 ± 39% of the preinduction control value 40 and 60 min after induction (n = 22; 10 layer 2–3 and 12 layer 5 neurons). In contrast, the same pairing protocol applied to synapses located at the distal half of basal branches failed to evoke LTP (Fig. 1). In fact, a tendency for long-term depression (LTD) was observed. The average EPSP amplitude at these distal locations was 91 ± 7 and 85 ± 9% of the preinduction control value 40 and 60 min after induction (Fig. 1) (n = 14; 6 layer 2–3 and 8 layer 5 neurons; in eight cases, measurements were performed from the proximal and distal regions of the same branch; p = 0.06 for comparison of the amplitudes obtained 40 and 60 min after induction to the preinduction control value). A similar difference between synapses located proximally and distally was observed with two additional induction protocols: first, pairing a burst of BAPs with a train of four EPSPs following Markram et al. (1997a) (n = 5; 94.1 ± 11.4% for distal synapses and 178 ± 33% for proximal synapses 40 min after induction) and second, theta-burst stimulation (n = 4; 97.3 ± 14.3% for distal synapses and 179 ± 48% for proximal synapses 40 min after induction; for more details of stimulation protocols, see Material and Methods). In addition to the focal stimulations, we attempted to evoke LTP by stimulating a broader region of the proximal and distal basal tree. To achieve a broader activation, we positioned an extracellular tungsten electrode 50–100 μm distal to the outer border of the basal tree (for distal activation) and 50 μm away from the soma (for proximal activation). Similar to focal stimulation protocols, we observed LTP proximally and not distally (n = 5; 92.6 ± 21.4% for distal synapses and 198 ± 38% for proximal synapses 40 min after induction).
In our experiments, the intracellular solution contained OGB-1 or OGB-6 (200 μm) for visualization and localization purposes. These calcium-sensitive dyes introduced extra calcium buffering capacity to the cell. To control for this variable, we replaced the calcium-sensitive dye OGB with Alexa 488, which has no calcium-buffering capacity. No significant differences were observed between experiments with OGB or Alexa dyes in the pipette. When Alexa was present in the pipette 40 min after induction, the average EPSP amplitude was 193 ± 45% proximally and 94 ± 9% distally (n = 5; p > 0.3 compared with OGB experiments).
Most papers in the literature that examined LTP induced by pairing protocols in basal dendrites did not specify the location of activated synapses. However, paired recordings experiments that performed detailed anatomical reconstructions reported that the activated synapses that underwent LTP were mostly located in the proximal portions of basal dendrites (Markram et al., 1997a,b). Thus, our findings regarding proximal basal dendrites are consistent with data in the literature, whereas the literature holds no specific information with regard to synapses located at distal basal dendrites.
Calcium transients in proximal and distal basal dendrites of layer 5 and layer 2–3 neurons
Previous studies have shown that induction of LTP is closely linked to the local calcium transients evoked during the induction period (Artola and Singer, 1993; Bear and Kirkwood, 1993; Bliss and Collingridge, 1993; Franks and Sejnowski, 2002; Sjostrom and Nelson, 2002). Thus, we compared the calcium transients evoked by BAPs, EPSPs, and pairing of both in proximal and distal regions of basal dendrites. To minimize dye saturation problems, we performed calcium imaging measurements with high concentration (0.5–1 mm) of the low-affinity dye OGB-6. We found no significant differences in the calcium transients evoked by the three protocols between proximal and distal regions of the same basal dendrite (Fig. 2). On average, calcium transients (ΔF/F) evoked by the LTP pairing protocol were 110 ± 58% in eight proximal dendrites and 120 ± 57% in 12 distal dendrites (p > 0.5). The similarity in the calcium transients in proximal and distal basal dendrites suggested that differences in calcium concentrations were unlikely to account for the inability of inputs in distal basal dendrites to undergo LTP. However, we cannot rule out the possibility that differences in the amplitude or time course of local subcellular calcium concentrations play a role in this phenomenon. It is interesting to note that, in contrast to basal dendrites, in apical dendrites the BAPs induced calcium transients decrease significantly with distance (Spruston et al., 1995; Larkum et al., 1999b).

Calcium transients in proximal and distal basal dendrites. A, Experimental setup. A basal branch of a layer 5 pyramidal neuron loaded with OGB-6 (1 mm). Calcium imaging was performed from a proximal (red) and a distal (blue) region. Calcium imaging measurements were collected from dendritic spines both in the proximal region (dotted red line) and the distal region (dotted blue line). B, Calcium transients presented as ΔF/F (%) were evoked for proximal (red) and distal (blue) regions by local EPSPs (bottom left), a train of four BAPs (bottom middle), and pairing of a train of four BAPs with local EPSPs (bottom right). C, A summary plot of the peak calcium transients at proximal and distal regions evoked by a train of BAPs, EPSP, and pairing of a train of BAPs and EPSP (as in the induction protocol) is presented. All measurements were done with OGB-6 (0.5–1 mm). Note that no significant difference in the calcium transients was observed between proximal and distal regions in all stimulus paradigms used (p values were not significant for all three comparisons). Error bars represent SD.
NMDA spikes are insufficient to induce LTP in basal dendrites of layer 5 and layer 2–3 pyramidal neurons
Dendritic spikes can serve as an alternative postsynaptic signal for the induction of LTP (Golding et al., 2002). We next investigated whether NMDA spikes can serve as the postsynaptic signal for LTP in distal basal dendrites of layers 5 and 2–3 pyramidal neurons. Because NMDA spikes have only been reported previously for layer 5 neurons, we first characterized these spikes in basal dendrites of layer 2–3 neurons as well. Similar to layer-5 pyramidal neurons (Schiller et al., 2000; Polsky et al., 2004), coactivation of spatially clustered synaptic inputs in basal dendrites of layer 2–3 pyramidal neurons initiated local NMDA spikes (Fig. 3A,B). NMDA spikes in layer 2–3 pyramidal neurons had an average somatic threshold of 2.94 ± 0.74 mV, average amplitude of 11.06 ± 5.01 mV, and average half-width of 80.4 ± 38.3 ms (n = 12). Similar to layer 5 neurons, NMDA spikes in layer 2–3 neurons evoked large local calcium transients (Fig. 3C). On average, the local calcium transients evoked by NMDA spikes were approximately sixfold larger than those evoked by a train of BAPs (n = 9; 43 ± 20 and 254 ± 44% for calcium transients evoked by a train of four action potentials and NMDA spikes in distal basal dendrites, respectively). Similar to layer 5 pyramidal neurons (Schiller et al., 2000), glutamate uncaging onto basal dendrites of layer 2–3 neurons showed that NMDA spikes could be initiated in the presence of voltage-gated sodium and calcium channel blockers (TTX 1 μm, nickel 100 μm, cadmium 100 μm), whereas application of the NMDA receptor blocker APV (50 μm) blocked the spike altogether (Fig. 3D).
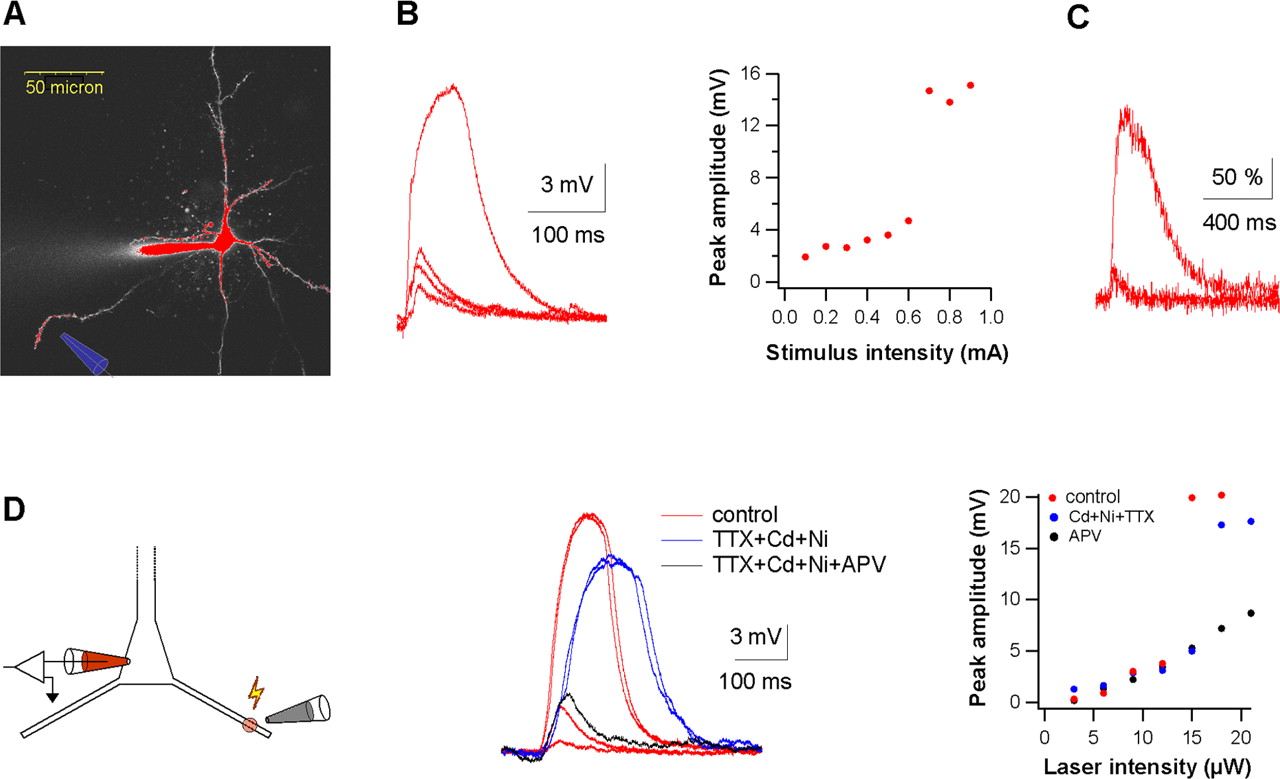
NMDA spikes in basal dendrites of layer 2–3 pyramidal neurons. A, Experimental set up: a layer 2–3 pyramidal neuron was loaded with OGB-6 (500 μm), and a theta-stimulating electrode (shown schematically) was placed close to a distal region (120 μm from the soma). B, Postsynaptic voltage responses evoked by two stimuli at 100 Hz are shown for different stimulus intensities. The stimulus intensity was increased gradually until an all-or-none local spike was initiated. The peaks of the postsynaptic voltage responses are plotted as a function of the synaptic stimulus intensity (right). C, Calcium transients obtained from an activated dendritic spine evoked by a local dendritic spike, a burst of four action potentials (100 Hz), and an EPSP (2 mV at the soma). D, NMDA spikes evoked by UV-laser uncaging of glutamate (Left, experimental set up shown schematically). Caged glutamate (MNI-glutamate, 5 mm) was delivered by pressure ejection locally through an electrode (2 μm tip diameter, 5 mbar). Middle, Single traces of the postsynaptic voltage response evoked by glutamate uncaging at increasing laser intensities in control conditions (red) and after addition of TTX (1 μm), cadmium (100 μm), and nickel (100 μm) (blue). Adding APV to the cocktail of blockers (50 μm; black) blocked the spike. Right, The peak postsynaptic amplitude is plotted as a function of the laser intensities for control conditions, in the presence of TTX, cadmium, and nickel (red) and in the presence of TTX, cadmium, nickel, and APV (black).
After establishing that basal dendrites of layer 2–3 pyramidal neurons also support the initiation of local NMDA spikes, we next investigated the possible role of NMDA spikes in the induction of LTP in both layers 5 and 2–3 basal dendrites. Surprisingly, pairing of EPSPs with NMDA spikes evoked by two focal theta electrodes 30–40 μm apart (typically 15–30 repetitions at 0.1 Hz, and four neurons with 60 repetitions) did not evoke robust LTP in both layer 2–3 and layer 5 pyramidal neurons (Fig. 4). On average, the EPSP amplitude 40 min after induction was 102 ± 44% of the preinduction control value (48 dendrites, 28 layer 2–3, 20 layer 5; no significant difference between layer 5 and layer 2–3 neurons). When we used a small number of repetitions in the induction protocol (one to five only), no change was observed in the EPSP amplitude, in contrast to the report by Holthoff et al. (2004). The difference between our findings and those reported by Holthoff et al. (2004) may result from differences in the experimental conditions including animal species (rats vs mice), brain area (somatosensory vs visual), and type of dendrites tested (basal vs a mixture of apical, oblique, and basal).

NMDA spikes fail to induce LTP in basal dendrites of neocortical pyramidal neurons. A, Induction protocol. NMDA spikes (2 stimuli at 50 Hz) were repeated 30 times at 0.1 Hz. B, Control individual response EPSP amplitudes were recorded (0.06 Hz) before (10 min) and after the induction protocol of NMDA spikes (50 min). C, Average unitary EPSPs before (blue) and after (red) induction protocol were identical. D, Summary data of 48 experiments presenting normalized average EPSP amplitude before and after the induction show no significant change. Error bars represent SEM.
The experiments involved with NMDA spikes were performed only on the distal half of basal branches, because we were unable to reliably obtain synaptically evoked NMDA spikes in proximal basal dendrites, despite our ability to stimulate these regions focally. Glutamate uncaging and modeling studies indicate that initiation of NMDA spikes in proximal basal regions requires much larger currents compared with distal regions (A. Polsky and J. Schiller, unpublished observations). We speculate that these current requirements were not satisfied in our experiments.
Our data show that, despite the fact that NMDA spikes were accompanied by very large calcium transients (Fig. 3C), they did not cause LTP. We raised the possibility that calcium influx evoked by NMDA spikes was too high to induce LTP. To examine this possibility, we attempted to induce LTP by pairing EPSPs and NMDA spikes in the presence of 4–10 mm intracellular BAPTA. Despite partially buffering the intracellular calcium concentrations, no long-term changes in the average EPSP were observed. Forty minutes after induction in the presence of intracellular BAPTA, the average EPSP amplitude reached 100 ± 52% of the preinduction control EPSP amplitude [n = 15 in eight neurons; no significant differences between layer 2–3 (n = 9) and layer 5 neurons (n = 6)].
Previous studies reported that the NR2B subunits of the NMDA receptor mediate LTD rather than LTP (Liu et al., 2004; Massey et al., 2004). We raised the possibility that during generation of NMDA spikes the NR2B subtype of NMDA-receptor subunits are preferentially activated and, hence, preclude induction of LTP. However, the selective NR2B subunit blocker ifenprodil (3 μm) reduced the amplitude of the basal spikes by <10% (n = 4). Moreover, we failed to induce LTP by pairing of NMDA spikes and distal EPSPs in the presence of ifenprodil (n = 3).
BDNF and NMDA spikes can mediate LTP in distal basal dendrites
Together, our findings indicate that basal dendrites contain two separate compartments with respect to the ability to undergo LTP. The first compartment, located at the proximal half of the basal tree, contains synapses that can undergo plasticity in response to the global suprathreshold activity of the neuron using pairing protocols. The second compartment, located at the distal half of the basal tree, contains synapses that are resistant to plasticity changes by either global BAPs or local dendritic spikes.
In light of these findings, we next investigated whether distal synapses still retain their ability to undergo LTP under more special conditions. More specifically, we examined whether these synapses can undergo LTP when a neuromodulatory molecule is present during the induction period. Because BDNF is a potent neurotrophic factor that modulates gating of NMDA-receptor channels (Levine and Kolb, 2000), and also enhances long-term plasticity (Korte et al., 1995; Kang et al., 1997; Black, 1999; Minichiello et al., 1999; Schuman, 1999; Kovalchuk et al., 2002; Du and Poo, 2004), we investigated its effect on plasticity of distal basal synapses.
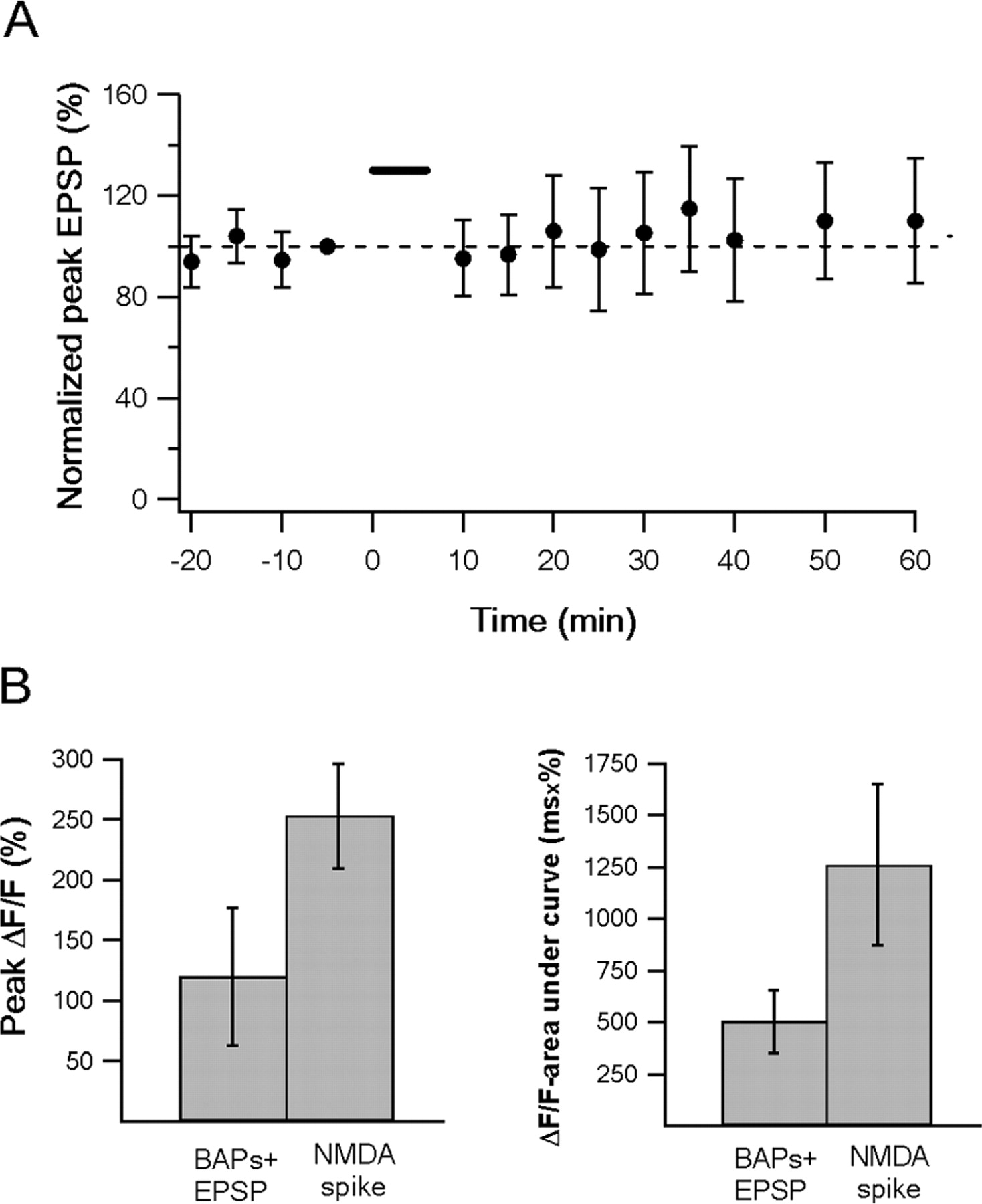
Pairing of BDNF with NMDA spikes and local EPSPs at the same dendritic segment caused a large and robust LTP (Fig. 5). On average, pairing of EPSPs and NMDA spikes (15 times at 0.1 Hz) in the presence of locally applied BDNF resulted in a 319 ± 116% increase of the EPSP amplitude, compared with its control value (n = 16, 10 layer 2–3, 6 layer 5; no significant difference between layer 5 and layer 2–3 neurons). In fact, five repetitions of paired NMDA spikes and EPSPs (0.1 Hz) were sufficient to induce LTP in the presence of BDNF (n = 4).

Pairing of NMDA spikes and BDNF is necessary for LTP induction in distal basal dendritic regions. A, Experimental setting. A layer 2–3 pyramidal neuron was loaded with OGB-1 (200 μm). Two stimulating electrodes (red) and a third electrode used to deliver BDNF (yellow; 50 ng/ml) were positioned near a basal dendrite. B, Peak EPSP amplitude before and after the induction protocol (black, voltage stimulation; green, BDNF application). The inset shows the induction protocol. NMDA spikes (15, 0.1 Hz) were delivered concomitant to local BDNF injection. C, A comparison of an average EPSP before the induction (10 min; black), after the induction (60 min; red), and in the presence of APV (blue) is shown. Note the large increase in the AMPA component. D, A summary plot of a normalized average of 16 similar experiments shows a large potentiation after this induction protocol (319 ± 116%). E, As a control to the requirement of NMDA spikes, the same protocol was repeated, but instead of evoking spikes, only local EPSPs were evoked (black, control; blue, 40 min after EPSP induction). Later, in the same dendrite, a second induction was performed with local NMDA spikes (red trace; 40 min after induction). The cell in B, C is different from the cell in E. Error bars represent SEM.
The effect of BDNF on LTP was mediated by the receptor tyrosine kinase TrkB pathway. Pretreatment of the slice for 10 min with the tyrosine kinase antagonist K252a (150 nm) markedly reduced the extent of LTP evoked by the pairing of EPSPs with NMDA spikes and BDNF. In all experiments, NMDA spikes were not affected by K252a. In the presence of K252a, the average EPSP amplitude measured 40 min after induction increased by only 154 ± 49% (n = 4), compared with 319 ± 116% without the tyrosine kinase blocker (p = 0.02).
Induction of LTP required the initiation of NMDA spikes. When EPSPs rather than NMDA spikes were paired with BDNF, no long-term synaptic plasticity was induced and the amplitude of EPSPs remained unchanged (Fig. 5E) (n = 4). Application of BDNF on inactive synapses also had no long-term effects on the EPSP amplitude (n = 3). In addition, when we activated only the NMDA-spike site (in the presence of BDNF) while the second electrode at the EPSP site (30–40 μm apart) was not activated during the induction protocol, no potentiation of the unactivated EPSP site was observed (n = 5).
We next investigated whether distal synapses can undergo LTP using pairing of EPSPs and BAPs in the presence of BDNF. Pairing of EPSPs with BAPs in the presence of BDNF failed to induce LTP in distal synapses (Fig. 6A). The average EPSP amplitude 40 min after pairing of BAPs and EPSP in the presence of BDNF was 105 ± 24% of the control value (n = 12). To further investigate the mechanism underlying the difference in potency to induce LTP between NMDA spikes and pairing of BAPs and EPSP, we compared the calcium transients evoked by both protocols. We found that NMDA spikes were associated with significantly larger and broader calcium transients compared with paired BAPs and EPSP (Fig. 6B) The peak and area under curve of calcium transients were 254 ± 44% and 1230 ± 388%/ms for NMDA spikes (n = 7) and 120 ± 57% and 503 ± 151%/ms for paired BAPs and EPSP (n = 12; p < 0.001 for both cases). We hypothesize that the difference between the calcium influx evoked by NMDA spikes and the pairing of BAP and EPSP can underlie the difference in their potency to mediate LTP in synapses located in distal basal dendrites. However, other possible mechanisms such as differential activation of NMDA linked biochemical cascades can also contribute to this phenomenon.

Pairing protocol in the presence of BDNF did not induce LTP in distal basal dendrites. A, Pairing BAPs with EPSPs (10 ms delay) in the presence of BDNF did not induce LTP in distal basal dendrites. The pairing protocol consisted of a burst of BAPs (4 BAPs; 100–150 Hz), delivered 10 ms after a single EPSP in the presence of local application of BDNF. A summary plot of a normalized average of 12 experiments using this protocol is shown. Error bars indicate SEM. B, Comparison of the averaged (mean ± SD) peak calcium transients (left) and area under the curve (right) evoked by pairing of BAPs and EPSP and NMDA spikes.
Together, our findings indicated that synapses innervating distal basal dendrites are relatively resistant to plasticity changes but still retain their ability to undergo LTP under special conditions. These synapses can undergo LTP when paired with local NMDA spikes in the presence of BDNF. Additional in vivo studies are required to determine whether and under what circumstances these induction conditions are fulfilled in the intact animal.
Temporal rules of NMDA spike-mediated LTP in basal dendrites
STDP opens a relatively narrow time window for potentiation and adheres to strict temporal rules with respect to the relative timing of presynaptic and postsynaptic activation ((Markram et al., 1997a; Bi and Poo, 1998; Feldman, 2000; Paulsen and Sejnowski, 2000; Sjostrom et al., 2001, 2002; Dan and Poo, 2004). We addressed the temporal rules governing NMDA spike-mediated LTP. In these experiments, we varied the time delay between NMDA spikes and the paired EPSPs evoked by a second electrode located 30–40 μm along the same branch. The BDNF was locally applied throughout the induction protocol period. NMDA spike-mediated LTP showed a dramatically different temporal window than STDP (Fig. 7). First, only LTP and not LTD was evoked. Second, NMDA spikes opened a prolonged time window for plasticity, lasting ∼150 ms. Third, LTP was induced regardless of whether spikes preceded or followed EPSPs. However, the potentiation time window was asymmetrical and preferred postsynaptic before presynaptic activation order at a ratio of 5:1 (Fig. 7C).

A large time window for plasticity after induction with NMDA spikes. Two stimulating electrodes were positioned (40 μm apart) at a basal dendrite. The time delay between an EPSP evoked with one electrode and a spike evoked with a second electrode was changed from 50 ms (EPSP before spike) to −200 ms (EPSP after spike). All inductions were performed in the presence of BDNF delivered locally with a third electrode. A, Example experiment presenting average peak in control and after induction protocol using a time delay of 20 ms (inset, EPSP before spike). B, Same as A, except that the time delay was −100 ms (inset, EPSP after spike). C, Normalized average summary before and after induction at delays from 50 ms to −200 ms from 42 experiments. Note the large time window of ∼150 ms opened by the NMDA spike for plasticity. Error bars represent SEM.
Discussion
In this study, we report that the basal tree is composed of proximal and distal plasticity compartments. Inputs in the proximal compartment undergo plasticity using conventional pairing protocols. In contrast, inputs in the distal plasticity compartment are relatively resistant to change and require unusual conditions to do so. These synapses undergo LTP only when NMDA spikes are combined with a gating molecule (BDNF). It is possible that plasticity rules change gradually between the proximal and distal compartments with no sharp borders between the two compartments. Additional experiments with finer spatial resolution of focal activation will be needed to clarify this point.
Location-dependent plasticity rules were reported previously for apical dendrites of layers 2–3 and layer 5 pyramidal neurons (Froemke et al., 2005; Sjostrom and Hausser, 2006). Our findings indicate that plasticity compartments markedly differ between basal and apical dendrites. In apical dendrites of layers 2–3 pyramidal neurons, proximal and distal inputs can undergo LTP when paired with BAPs and differences between proximal and distal inputs are mainly manifested in a slight amplitude reduction of LTP (Froemke et al., 2005). In addition, it was shown previously that distal apical dendrites of layer 5 pyramidal neurons undergo LTD and not LTP in response to STDP induction protocols (Letzkus et al., 2006; Sjostrom and Hausser, 2006). It is important to note that distal apical dendrites of both layer 5 and CA1 pyramidal neurons undergo LTP in response to the local spikes within the dendritic branch (Golding et al., 2002; Lisman and Spruston, 2005) (J. Schiller, unpublished observations regarding apical dendrites of layer 5 pyramidal neurons). In contrast to apical dendrites, inputs innervating distal basal dendrites fail to undergo LTP when paired with either BAPs or local spikes within the branch.
The significance of functional dendritic compartmentalization with respect to plasticity and integration depends on the spatial dendritic innervation pattern (Hausser and Mel, 2003). Previous studies have reported spatial segregation of inputs carrying different information into different dendritic regions (Single and Borst, 1998; Megias et al., 2001; Euler et al., 2002). For example, feedback corticocortical inputs terminate on the tuft apical branches (Pandya and Yeterian, 1985; Cauller and Connors, 1994). Also, in basal dendrites, some evidence supports spatial dendritic segregation of inputs according to the information they carry. Inputs originating from within the same cortical column preferentially innervate proximal basal dendrites (Markram et al., 1997a; Feldmeyer et al., 2002). Interestingly, noradrenergic innervation may also differ along the basal dendritic tree (Benavides-Piccione et al. 2005). Thus, it is possible that the ability of different anatomical pathways to undergo LTP will markedly differ depending on the dendritic plasticity compartment on which they terminate.
In this study, we describe a special case of synapses that require the presence of both NMDA spikes and the neuromodulatory molecule BDNF to undergo LTP. The involvement of the neurotrophic factor BDNF has been well documented in a variety of developmental processes such as survival and differentiation (Binder and Scharfman, 2004) as well as synaptic plasticity (Korte et al., 1995; Black, 1999; Minichiello et al., 1999; Schuman, 1999; Schinder and Poo, 2000). Previous studies have shown that the addition of exogenous BDNF can enhance plasticity (Kovalchuk et al., 2002; Du and Poo, 2004). Moreover, genetic deletion of the BDNF or TrkB genes impaired the ability of synapses to undergo LTP in the hippocampus, hence, demonstrating the physiological role of endogenous BDNF in LTP induction (Korte et al., 1995, 1996; Kang et al., 1997; Bramham and Messaoudi, 2005). It is interesting to note that, in contrast to previous studies, the BDNF in the case of distal basal synapses acts as an essential “gating” molecule and not just as a facilitatory molecule as shown for other pathways (Schuman, 1999).
One of the main questions raised by our findings is what physiological conditions will promote the release of BDNF in distal synapses? The factors that control the release of BDNF in the brain in vivo are not yet clear. Previous studies have shown release of BDNF from both axons and dendrites (Hartmann et al., 2001; Kohara et al., 2001) in an activity-dependent manner (Bonhoeffer, 1996; Rocamora et al., 1996; Altar and DiStefano, 1998; Fawcett et al., 1998; Kohara et al., 2001; Kojima et al., 2001; Lessmann et al., 2003; Aicardi et al., 2004; Adachi et al., 2005). Our results show that the induction protocols we used in the slice preparation were not efficient in releasing BDNF to distal basal synapses. It is possible that under in vivo conditions, other activation patterns or activation of additional pathways will supply a sufficient amount of endogenous BDNF that, when paired with NMDA spikes, will result in LTP induction in distal synapses.
The mechanism by which the combined NMDA spikes and BDNF exert their effects in our case is not known. It has been reported previously that BDNF acts postsynaptically by depolarizing dendrites and evoking local calcium transients in dendritic spines (Blum et al., 2002; Kovalchuk et al., 2002). In our case, the signal required for LTP induction was the NMDA spike, which to begin with is a large local voltage signal introducing very a large calcium influx postsynaptically. Thus, it is unlikely that the mechanism by which BDNF acts is by adding postsynaptic depolarization or increasing the calcium influx. Rather, we hypothesize that BDNF acts on one or more processes of the postcalcium biochemical cascade leading to LTP (Huang and Reichardt, 2003). In addition, the mechanisms underlying the preference of NMDA spikes over BAPs in LTP induction at distal synapses are not known. NMDA spikes are associated with a much larger activation of NMDA current compared with EPSPs or BAPs. Induction of LTP by NMDA spikes may result from the larger calcium influx evoked by NMDA spikes, or by more effective activation of NMDA receptor-linked biochemical cascades. In addition, NMDA spikes may be more efficient in promoting secondary activation of mGluRs compared with BAPs. Additional studies are required to differentiate between these different possibilities.
Presently, it is difficult to determine the physiological role of the distal plasticity compartment. It is possible that the special conditions required for LTP in distal basal dendrites are not fulfilled in vivo and inputs terminating on distal basal dendrites serve to carry hard-wired information that is protected from activity-dependent changes. If, however, NMDA-spike mediated LTP occurs in vivo, it can potentially serve three main roles.
First, the temporal plasticity rules used when NMDA spikes serve as the postsynaptic signal markedly differ compared with STDP temporal rules. The potentiation time window of NMDA spike-mediated LTP are much longer than that of STDP lasting ∼150 ms and, in contrast to STDP, a reversed pre/post activation order is preferred. The temporal rules of STDP are well suited to mediate plasticity of feedforward inputs. In contrast, the plasticity rules that apply to feedback synapses are expected to have different properties: they are expected to be “noncausal,” because feedback inputs will normally fire sometime after the neuron was activated. Moreover, the timing requirements for the induction of plastic changes should be much broader than for the feedforward pathway, given that feedback pathways are polysynaptic with varying numbers of intermediate neurons. The NMDA spike-mediated LTP provides all of these requirements; thus, it is well suited to potentiate feedback inputs. An interesting, yet speculative possibility is that feedback pathways preferentially terminate unto distal basal dendrites and, hence, are governed by a different set of plasticity rules.
A second possible role is in developmental wiring of the cortical network. Previous studies have shown that synaptic plasticity is age dependent. Certain pathways, most notably the thalamocortical fibers, lose their ability to undergo long-term synaptic plasticity as the animal matures (Crair and Malenka, 1995; Kirkwood et al., 1995), whereas other pathways require stronger postsynaptic signals for the induction of LTP with maturation (Pike at al., 1999). Our study was performed on juvenile (17–24 d of age) rats. It is possible that different plasticity rules serve to wire different anatomical pathways or that maturation of the plasticity rules of inputs depends on their dendritic location, with distal inputs maturing earlier. Additional studies are required to establish whether distal inputs can undergo plasticity with STDP rules in younger animals and whether proximal inputs require more intense activation to undergo plasticity in older animals.
A third potential physiological role of the NMDA spike-mediated LTP is to increase the stability of previously acquired information. Synapses located in the distal plasticity compartment are unlikely to change accidentally as a result of the ongoing network activity, because they require both the initiation of a local spike and the release of BDNF. The existence of different plasticity compartments with diverse plasticity rules may serve to increase the processing capabilities of the network (Bastian et al., 2004).
Footnotes
-
This work was supported by the National Institutes of Health, The Israel Science Foundation, and the Rappaport Foundation. We thank M. Hausser, M. London, B. W. Mel, Y. Poirazi, and Y. Schiller for their helpful comments on this manuscript.
- Correspondence should be addressed to Dr. Jackie Schiller, Technion Medical School, Bat-Galim, Haifa 31096, Israel. jackie{at}tx.technion.ac.il
References
- Adachi et al., 2005.↵
- Aicardi et al., 2004.↵
- Altar and DiStefano, 1998.↵
- Artola and Singer, 1993.↵
- Bastian et al., 2004.↵
- Bear and Kirkwood, 1993.↵
- Benavides-Piccione et al., 2005.↵
- Bi and Poo, 1998.↵
- Binder and Scharfman, 2004.↵
- Black, 1999.↵
- Bliss and Collingridge, 1993.↵
- Blum et al., 2002.↵
- Bonhoeffer, 1996.↵
- Bramham and Messaoudi, 2005.↵
- Cauller and Connors, 1994.↵
- Crair and Malenka, 1995.↵
- Dan and Poo, 2004.↵
- Du and Poo, 2004.↵
- Euler et al., 2002.↵
- Fawcett et al., 1998.↵
- Feldman, 2000.↵
- Feldmeyer et al., 2002.↵
- Franks and Sejnowski, 2002.↵
- Froemke et al., 2005.↵
- Golding et al., 2002.↵
- Hartmann et al., 2001.↵
- Hausser and Mel, 2003.↵
- Holthoff et al., 2004.↵
- Huang and Reichardt, 2003.↵
- Johnston et al., 2000.↵
- Kang et al., 1997.↵
- Kirkwood et al., 1995.↵
- Koester and Sakmann, 1998.↵
- Kohara et al., 2001.↵
- Kojima et al., 2001.↵
- Korngreen and Sakmann, 2000.↵
- Korte et al., 1995.↵
- Korte et al., 1996.↵
- Kovalchuk et al., 2002.↵
- Larkman, 1991.↵
- Larkum et al., 1999a.↵
- Larkum et al., 1999b.↵
- Lessmann et al., 2003.↵
- Letzkus et al., 2006.↵
- Levine and Kolb, 2000.↵
- Lisman and Spruston, 2005.↵
- Liu et al., 2004.↵
- Magee, 1999.↵
- Magee, 2000.↵
- Markram et al., 1997a.↵
- Markram et al., 1997b.↵
- Massey et al., 2004.↵
- Megias et al., 2001.↵
- Migliore et al., 1999.↵
- Minichiello et al., 1999.↵
- Pandya and Yeterian, 1985.↵
- Paulsen and Sejnowski, 2000.↵
- Pike et al., 1999.↵
- Polsky et al., 2004.↵
- Rocamora et al., 1996.↵
- Schiller et al., 1997.↵
- Schiller et al., 2000.↵
- Schinder and Poo, 2000.↵
- Schuman, 1999.↵
- Single and Borst, 1998.↵
- Sjostrom and Hausser, 2006.↵
- Sjostrom and Nelson, 2002.↵
- Sjostrom et al., 2001.↵
- Spruston et al., 1995.↵
- Stuart et al., 1997.↵





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}